BSc Microbiology Microbial Genetics Notes Study Material
BSc Microbiology Microbial Genetics Notes Study Material: BSc is a three-year program in most universities. Some of the universities also offer BSc Honours. After getting enrolled for BSc, there are certain things you require the most to get better grades/marks in BSc. Out of those, there are BSc Study Material, BSc Sample Model Practice Mock Question Answer Papers along with BSc Previous Year Papers. At gurujistudy.com you can easily get all these study materials and notes for free. Here in this post, we are happy to provide you with BSc 2nd Year Microbiology Microbial Genetics Notes Study Material.

BSc Microbiology Microbial Genetics Notes Study Material
During the last twenty-five years or so, there has been a dramatic increase in our knowledge of the mechanisms that determine the characteristics of microorganisms. What we know indeed of the structure and organization of DNA, has been possible only through the work on microbial genetics, particularly of bacteria and fungi. Microbes have proved ideal organisms of study in molecular biology.
We now understand the functioning of the genome at the molecular level and make efforts to manipulate cells for improving their specific desired properties. This has resulted in a new phase of exploiting bacteria and fungi in industry i.e. in biotechnology.
The hereditary characteristics are coded on DNA, in discrete units – genes. During cell division, DNA replication occurs, for which decoding of the genes is carried out in two steps called transcription and translation giving rise to the polymers of RNA and protein respectively. This information transfer in cells almost always occurs in the way.
Gene
The important relationship between DNA and the characteristics of an organism was derived from studies of both fungi (Neurospora crassa) and bacteria (Pneumococcus, E. coli). The main finding that genes act by controlling the specificity of protein synthesis was obtained from work with mutants of the fungus Neurospora crassa, which led to the proposal that
1 gene = 1 enzyme

Bacteria, particularly E.coli have been used to define this relationship more precisely. Thus:
1 gene (or cistron) = 1 polypeptide
This definition reflects the discovery that not all polypeptides are enzymes. Some play a structural role (as flagellin), and others are regulatory (as the catabolite repressor protein, CRP).


Transcription is the copying of DNA to give an intermediary molecule, the mRNA, carried out by RNA polymerase. In translation, the RNA is translated into a sequence of amino acids that make the polypeptide. This requires rRNA and tRNA. Translation takes place in the ribosome.
Control of Transcription
There are two types of genes: structural and regulatory.
- Structural genes. These are the majority of genes, which produce specific polypeptides which may act directly or in combination with other peptides to form enzymes or structural proteins. Such genes are called structural genes. In bacteria, these genes usually occur as a cluster on the chromosome, the region is called an operon. Such multigenic operons produce a single molecule of mRNA initiated by the binding of RNA polymerase to a promoter sequence at one end of the operon.
- Regulatory genes. These are situated immediately adjacent to the promoter sequence, sometimes even overlapping with it, and are the regulatory sequences that control the initiation of transcription. These sequences are the site of action of polypeptides produced by regulatory genes.

There are basically two types of control of transcription, negative and positive.
(a) Negative control. The basic action of these regulatory polypeptides is to combine within or close to the promoter at a region known as the operator. The binding of the regulatory protein to the operator prevents the binding of RNA polymerase to the promoter. Synthesis of RNA is thus inhibited and no synthesis of the protein coded for by the operon occurs. In other words, control is essentially by competition between the RNA polymerase and the regulatory protein for the promoter. (BSc Microbiology Microbial Genetics Notes Study Material)
(b) Positive control. This type of control has been recognized only during the last ten years. Here the initiation of transcription is dependent upon the binding of the regulator to the promoter. Examples are many, such as (i) the catabolite repressor protein (CRP) where CRP forms an active complex with cyclic AMP (CAMP), which acts on many different operons to increase the rate of their transcription (over 200 genes are regulated by CRP-CAMP complex in E. coli). (BSc Microbiology Microbial Genetics Notes Study Material)
This is called the global regulatory system as it affects genes of many different metabolic systems. (ii) specific regulator, the one which is specific to only one operon or group of operons. When a number of operons are under the control of a single specific regulator it is termed a regulon. An example of this type is the arabinose regulation of E. coli. (BSc Microbiology Microbial Genetics Notes Study Material)
Regulation of gene expression usually requires the presence of another molecule other than the protein produced by the regulator gene. This is usually either the inducer or the repressor depending on its mode of action and the processes of regulation are known as induction or repression respectively. Both can be found in either negative or positive regulatory systems.
- Induction. The inducer specifically combines with the regulatory protein rendering it either effective at binding to the regulatory sequence and thus stimulating transcription (positive regulation) or incapable of acting on the operator sequence and thereby switching on induced protein synthesis (negative regulation).
- Repression. The binding of the repressor to the regulatory molecule either prevents interaction of the positive regulator with the regulatory sequence or increases the affinity of the negative regulator for the operator sequence (negative regulation) and thus stops transcription.
Mutations in Microorganisms
The mutation is a natural phenomenon resulting in variations within any population of cells. This is a change in the DNA sequence of a gene and is said to lead to a change in the genotype of the organism. Various types of mutants are found in microorganisms. These are as follows:
- Auxotrophic mutants. Many microbes such as E. coli can grow on a medium containing a single carbon and energy source. They are called prototrophs. If a mutation occurs which results in the loss of the ability to synthesize an essential metabolite such as an amino acid or a growth factor, it will express itself in a nutritional requirement for that substance. Such mutants are called auxotrophs. These proved much useful in the establishment of the relationship between genes and enzymes, as markers for genetic experiments in the construction of genetic maps, etc. (BSc Microbiology Microbial Genetics Notes Study Material)
- Resistant mutants. Bacteria may develop resistance to antibiotics and phages spontaneously through a range of mechanisms, e.g. loss of cell surface components that act as phage receptors, and acquisition of enzymes able to metabolize the antibiotic.
- Metabolic mutants. These mutants have lost the ability to use a particular carbon source and are usually affected either in transport or metabolism. The mutants which have lost the capacity to make specific cell components as the capsular polysaccharides and some of those which exhibit altered colonial shape (arising from mutations affecting cell wall synthesis) also belong to this category.
- Regulatory mutants. In these mutants, the mutation affects either the regulatory region of the promoter of the gene or the activity of a regulatory protein.
The above-mentioned types of mutation are common in nature and the laboratory. This classification of mutations is based on their effects on the genotype of the cell. There are also another different kind of mutations, which are shown in Table.

Spontaneous mutations. They take place in nature without human intervention or identifiable cause. On average in a colony of one billion (10%) bacteria, at least one mutant may be present. Since 1976, a penicillin-resistant strain of Neisseria gonorrhoeae (gonorrhea-causing bacterium) has been emerging in human populations. Antibiotic-resistant serotypes of Salmonella are also developing in nature.
Induced mutations. These are the mutations in which the cause can be identified. More often they result from planned experiments, in which microbes are subjected to physical or chemical agents. The most common agents used to induce mutations (mutagens) are ultraviolet light, nitrous acid, and several base analogs (the chemicals closely related to nitrogenous bases of DNA). One base analog is 5-bromouracial. Other mutagens include benzopyrene (in industrial soot and smoke), aflatoxin (a fungal toxin found in animal products and foods), etc.
Transposable Genetic Elements
Transposable genetic elements (TGE) are fragments of DNA. These may cause totally different types of mutations in nature. Two types of TGE are known: insertion sequences and (ii) transposons.
Insertion sequences. They are small segments of DNA with about 1000 base pairs, found at one or more sites on the bacterial chromosome, and appear to have no specific genetic information except their ability to insert onto a chromosome. Insertion sequences form copies of themselves, and the copies move from the normal positions into areas of gene activity. Here they interrupt the coding sequence.
Transposons. They are the so-called jumping genes for which Barbara Clintock won the 1983 Nobel Prize in Physiology or Medicine. Earlier in 1951, her work on Indian corn plants revealed that genes may not be always fixed elements and found in the same position on the same chromosome. She could describe that genes apparently moved from one chromosome to another. She watched over the changes in colour in her plants and the pigment genes responsible for colour change appeared to be switched on or off in particular generations.
More remarkable was that these switches occurred in later generations at different places along the same chromosome. In modern molecular genetics, these controlling elements are a two-element system: an activator gene and the other a dissociation gene. The activator gene can direct a dissociation gene to jump along the arm of the ninth chromosome in maize plants where colour is regulated.
The Jumping gene is identical to the transposon found in bacteria. Transposons, first identified and named in 1974 by British Microbiologists, R.W. Hedges and A.E. Jacobs, are larger than insertion sequences and carry information for protein synthesis. They contain genes for antibiotic resistance. They may move from plasmid to plasmid, from plasmid to chromosome, or from chromosome to plasmid. The movement of transposons is nonreciprocal i.e. an element moves away from its location and nothing takes its place. This contrasts with insertion sequences where copies move. (BSc Microbiology Microbial Genetics Notes Study Material)
Overlapping genes
For years scientists assumed that a mutation gene affected the production of a single protein. However, Frederick Sanger’s (1977) work indicated otherwise. He determined the entire nucleotide sequence of a viral DNA molecule and mapped its, 5374 bases. Analysis of the DNA by Sanger’s coworkers showed that at least four of the genes were overlapping. This showed that an individual triplet codes for two different amino acids. In 1958 Sanger won the Nobel Prize in chemistry for his sequencing of the amino acids in insulin. In 1980 he shared a second Nobel Prize in chemistry for mapping the bases of viral DNA.
Ames test
It was seen that about 90% of the agents that cause cancer in humans also cause mutations in bacteria. Working on these lines, Bruce Ames at the University of California, USA developed a procedure to determine whether a chemical can induce a bacterial mutation and thus be a potential agent of cancer. The procedure, the Ames test is relatively inexpensive, accurate, and rapid. A histidine-requiring strain of Salmonella typhimurium is inoculated into a plate containing the culture medium lacking histidine.
Normally the Salmonella strain will not grow as genes for histidine synthesis are lacking. Now the potential cancer agent is added to the medium and the plate is incubated. If a bacterial colony appears, one may conclude that the agent mutated the bacterial genes to code for enzymes needed for histidine synthesis. The agent is, therefore, a possible cause of cancer. If bacterial colonies fail to appear, no mutation takes place. However, the test works only within specified limits.
Enrichment and replica plating
Mutation rates can be increased by the use of mutagens like UV light, ionizing radiation, and chemicals like nitrous acid, nucleotide analogs, etc. Methods have been developed for the isolation of a particular desired type of mutant. There may be some difficulty in finding a suitable method to enrich specifically for the desired mutant and to recognize and isolate it when it may still be only a small proportion of the irradiated population. Direct selection can only be achieved when the mutation gives rise to resistance to a chemical or virus.
The enrichment step is very useful in increasing the percentage of cells carrying the mutated gene. One such method is penicillin (or more frequently ampicillin) enrichment. The cells are first grown in normal growth conditions (as those used for parent cells). The mutagenized cells are then transferred to an environment in which the desired mutant can not grow and after a short time (to allow the growth of the mutant to cease) ampicillin is added to kill any cells which are still able to grow. The survivors of such a treatment are enriched for the desired mutant. (BSc Microbiology Microbial Genetics Notes Study Material)
Nutritional and various other types of mutants are often detected by the replica plating techniques, developed by Joshua and Esther Lederberg in 1952 in order to provide direct evidence for the existence of preexisting mutations. Their actual experiment was concerned with replicating master plates of sensitive cells to two or more plates containing either streptomycin or phage.
When the replicas were grown, they were compared to the location of colonies on the master plate and any resistant colonies that appeared in the same position on all the replica plates were marked. The area of the master plate corresponding to the marked areas was cut out and the bacteria on it resuspended in a liquid medium. (BSc Microbiology Microbial Genetics Notes Study Material)
This method has been applied in numerous experiments to identify the occurrence of mutations. Many of the biochemical pathways in microbes were elucidated in this way by using nutritional mutants. Replica plating allows the observation of microbes under a series of growth conditions. In replica plating, a piece of sterile velvet is touched to the surface of an agar plate containing surface bacterial colonies. The fibers of velvet act as fine inoculating needles, picking up the bacterial cells from the surface of this master plate. The velvet with its attached microbes is then touched to the surface of a sterile agar plate, inoculating it.

In this manner, microbes can be repeatedly stamped onto media of differing compositions. The distribution of microbial colonies should be exactly replicated on each plate unless the colonies represent different genetic strains. Should a colony that develops on a complete medium fail to develop on a minimal medium that lacks a specific growth factor, the occurrence of a nutritional mutant is indicated. The microbes that do not grow on the minimal medium represent auxotrophic strains. (BSc Microbiology Microbial Genetics Notes Study Material)
This technique was specifically developed to answer an important and controversial question-did mutations occur spontaneously, or directed by the selective agent? For example in the case of penicillin resistance, does penicillin direct the mutation, or does it simply select out a naturally occurring spontaneous mutant? It was shown by statistical analysis of mutants that the process is indeed a spontaneous one. This method also allows the isolation of penicillin-resistant mutants without ever exposing the cells directly to penicillin.
Mutations and adaptability
The selection of spontaneous mutants by the environment and the exchange of plasmids (we shall refer to these DNA molecules later in this chapter) between organisms can occur more quickly in microorganisms. This may result in a rapid change in the genotype of the dominant organism in a population of cells. As compared with higher organisms, the mutation rates in microbes are higher and the spread of newly acquired characteristics occurs at a faster rate. This is due to the following:
(1) Most microbes are haploid, thus mutations cannot be masked by the allelic genes.
(2) They are unicellular, so many mutated cells can give rise to a new evolutionary line.
(3) Microbes can occur in high densities in restricted environments, where a mutant may have a selective advantage.
(4) Microbes grow very rapidly.
(5) Plasmid transfer is common, thus new traits do not have to be acquired by all organisms through a long process of mutation and evolution.
Due to the above-said characteristics, microbes, particularly prokaryotes, are able to adapt themselves to a wide variety of extreme environments.
Recombination in Prokaryotes
We have seen that genetic changes due to mutations can result in the acquisition of new biological characteristics and thereby allow evolutionary change. However, the evolution of the fittest organism in a particular environment can be enhanced if the transfer of genes between organisms is made possible by genetic recombination.
As compared with eukaryotes, where sexual recombination is of ordered nature, in prokaryotes the process is less well developed. It does not involve a true fusion of male and female gametes to produce a diploid zygote Instead there is the transfer of only some genes from the donor cell to produce a partial diploid. This is followed by recombination to restore the haploid state. There are three mechanisms by which these DNA fragments can pass from a donor to a recipient cell (i) transformation, (ii) transduction, and (iii) conjugation.
[I] Transformation
The transformation was discovered by an English bacteriologist, Frederick Griffith in 1928, who made a series of experiments with laboratory mice and two types of pneumonia-causing bacterium, Diplococcus pneumoniae. This bacterium has two types of strains. One type has smooth (S), capsulated cells, whereas another type has rough (R) non capsulated cells. The disease is caused by smooth-type of cells only i.e. smooth-type cells are pathogenic (virulent) whereas rough-type cells are harmless or nonpathogenic (avirulent).
The experiments conducted by him are illustrated. As shown in the figure, when live, harmless (rough type) cells were injected into the body of mice, the animal remained healthy. The injection of dead, pathogenic (smooth-type) cells into the body of mice also did not cause any disease. In a classic experiment, Griffith mixed live, harmless (rough-type) cells with the dead remains of pathogenic (smooth-type) cells, and injected the mixture into the laboratory mice. The live cocci taken in the mixture were encapsulated and formed rough colonies (R) on agar. The dead cocci taken in the mixture originally had a capsule and were taken from smooth (S) colonies on agar.
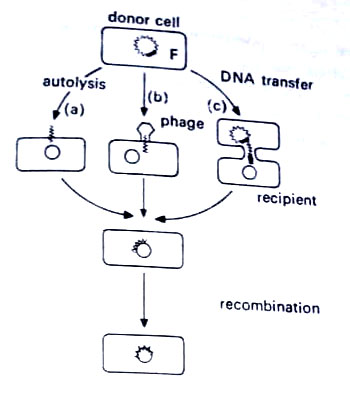
To Griffith’s surprise, the mice developed pneumonia and died. On autopsy (examination of tissue of dead animal), he isolated live, capsulated cells that formed smooth colonies on agar. Apparently, the live, harmless rough cocci had been transformed in the mice into live, pathogenic, smooth cocci.

A rough-to-smooth conversion (R → S) had been accomplished. Five years later, James L. Alloway of the Rockefeller Institute confirmed Griffith’s work using fragments from the dead smooth-type cells to transform the rough-type cells. In 1944, Oswald T. Avery, Colin M. MacLeod, and Maclyn N. McCarty, also of Rockefeller Institute found that deoxyribose-nucleic acid (DNA) isolated from the fragments could induce the transformation. At that time, DNA was an obscure chemical with little significance. The work of Avery, MacLeod, and McCarty helped bring it to the force. Their experiments were the first proof that in living organisms genetic matter is DNA.
The possible mechanism of transformation is shown in Figure. Though it takes place in less than 1% of a population, transformation is an important method of recombination in bacteria. A number of donor cells break apart and an explosive release and fragmentation of DNA follow. A segment of double-stranded DNA containing about 10-20 genes then passes through the cell wall and membrane of a recipient cell. Only a few competent recipient cells can take up the DNA.
After entry into the cell, an enzyme dissolves one strand of DNA leaving the second strand to be incorporated. This strand then displaces a segment from a strand of the recipient’s DNA. The displaced DNA is dissolved by another enzyme in the cell. The cell is now transformed. It will display its own traits as well as those coded by the new DNA. (BSc Microbiology Microbial Genetics Notes Study Material)

Transformation may also take place through the incorporation of plasmids into competent cells. In this case, no DNA is displaced. Rather, the plasmid adds genes to those already in the cell and multiplies along with the cell.
Since the 1940s, the transformation has also been demonstrated in species of Neisseria, and Bacillus. Haemophilus, Azotobacter, and Streptococcus. The process involves the transfer of DNA from the fragments of donor cells into the cytoplasm of a live recipient cell. Sections of single-stranded or double-stranded DNA may be taken up but only a single strand will align with the bacterial chromosome and becomes incorporated into it.
Transformations in bacteria have been observed in the ability to form a capsule, drug resistance and pathogenicity, and in nutritional patterns. Transformations are not common, however, because the large fragments of DNA molecule can not pass through the recipient’s cell wall or membrane. In nature, transformation may increase the pathogenicity of an organism.
In cells of some genera of bacteria, DNA binds to the cell surface before uptake. However, this is restricted only to some genera. This problem has been overcome in both, pro- and eukaryotes by the removal of the cell wall to form protoplasts. The addition of a compound fusion allows the protoplasts to fuse and mix of DNA of two or more cells for recombination. This technique is known as protoplast fusion.
When DNA is included in the incubation mixture transformation occurs simultaneously with fusion and thus small fragments of DNA e.g. plasmids can be introduced into the cells. Protoplast fusion can also be managed between members of different species by mixing their protoplasts in the presence of a fusinogen. In this way, the genetic exchange can be achieved between organisms for which no other gene transfer system is known to exist. (BSc Microbiology Microbial Genetics Notes Study Material)
[II] Conjugation
It involves the transfer of genetic material from one living cell to another during a period of contact. Whereas in both transformation and transduction any cell can act as a donor or a recipient, in conjugation some are donors of DNA while others are recipients. This process was first postulated by Joshua Lederberg and Edward Tatum in a series of experiments in 1946. Two strains of Escherichia coli were used.
One was unable to synthesize an essential compound A whereas the owner could not synthesize an essential compound B. Neither strain was able to grow in a culture medium lacking both, compound A and compound B because each strain lacked an important enzyme system.
Lederberg and Tatum mixed the cells of two strains, and after a short incubation time, placed samples on the medium lacking both A and B. Surprisingly, bacterial growth appeared in this medium. When a few colonies appeared, it was first thought that transformation had occurred. However, it was shown experimentally that physical cell contact was necessary for the recombination to take place.
Apparently, the genes for the synthesis of compounds A and B passed between the cells and yielded a recombined chromosome that could produce both the missing compounds. Their experiments are illustrated in Figure. Lederberg and Tatum shared the Nobel Prize of 1958 for their work in bacterial genetics. (BSc Microbiology Microbial Genetics Notes Study Material)

Later experiments by Francois Jacob and Elie L. Wollman established that bacteria were of two mating types. Certain male types or F+ or donor cells were those that donated some of their DNA, whereas female type, or F– or recipient cells, were the recipient of the genes. It was noted that recipient cells would rapidly become donor cells when certain small amounts of DNA were passed to a recipient cell.
This eventually led William Hayes to discover genetic factors, called fertility factors or F-factors, in the cytoplasm of the donor cell (male) apart from the chromosome. These are also called sex factors or F-plasmids. In contemporary microbiology, the F-factors are known to be types of plasmids. They are double-stranded loops of DNA, apart from the chromosome.

The factors contain about 20 genes, most of which are associated with conjugation. One function of these genes is to form a conjugation bridge or sex pilus, between donor and recipient cells. The F-factor then prepares for replication by the rolling circle mechanism. There is no complete process of replication but as in replication, the two strands begin to separate from each other and a single strand of the factor passes through the sex pilus to the recipient. When it arrives, enzymes synthesize a complementary strand, and a double helix forms again.
The double helix bends into a loop and the conversion from F to F+ cell is completed. Meanwhile, back in the donor cell, a new strand of DNA forms to complement the leftover strand of the factor. The word episome is commonly applied to plasmids that function in conjugation processes. (BSc Microbiology Microbial Genetics Notes Study Material)
High frequency of recombination. In the above-mentioned process, we have seen that the transfer of F-factors involved no activity of the bacterial chromosome. Therefore the recipient cell does not acquire new genes other than those on the F-factor. However, there exists in bacteria a type of conjugation that accounts for the passage of chromosomal material.
Strains of bacteria that display this ability are called the high frequency of recombination, or Hfr strains. Such strains were discovered in the 1950s by William Hayes in E. coli. Such strains developed when a male (F+) mutated to a super-male which showed a high frequency of recombination with a female (F–), hence the mutant called Hfr.